Glamdring, the Astrophysics compute cluster
 |
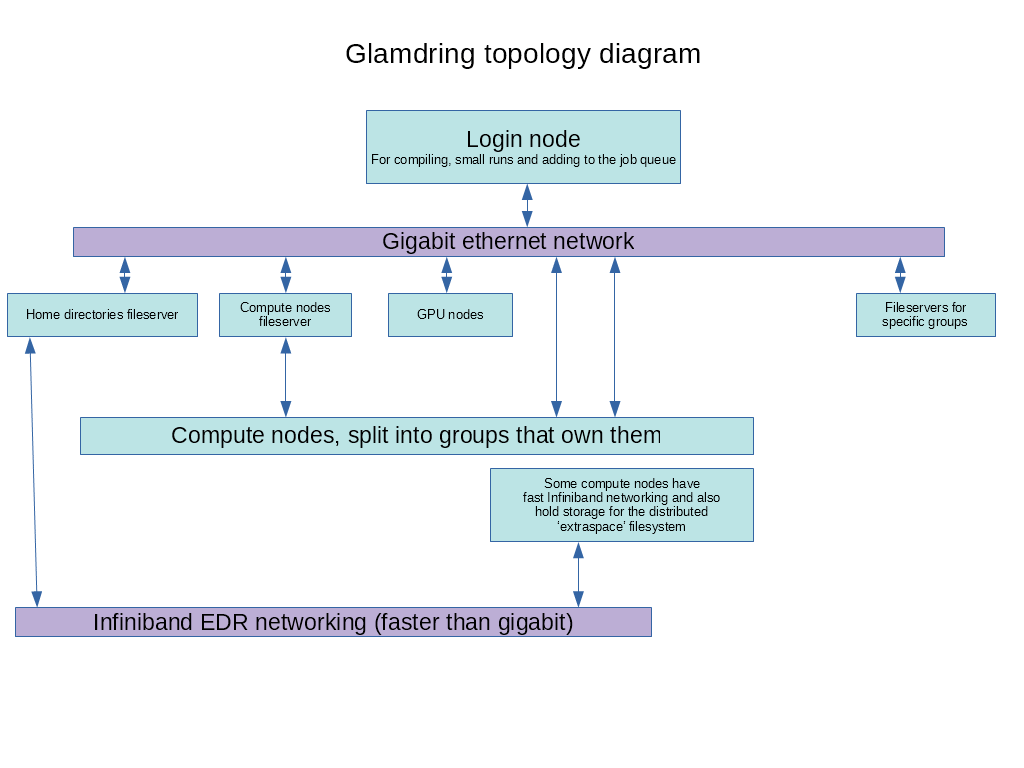
Glamdring is a linux-based, computing cluster consisting of 94, dual-cpu, multi-core, Intel-CPU based machines for people in Astro who need large amounts of CPU time. So we have a total of 1,200 CPU cores on which to run programs, either MPI-based, OpenMP based or normal "serial" programs. There is a topology map of the cluster HERE if you are interested. You can connect via SSH or x2go (a virtual desktop) or RDP (a virtual desktop). Please email jonathan.patterson@physics.ox.ac.uk if you want to connect via RDP - I have to enable it for you. RDP needs to be tunnelled through ssh or you need to connect via the Physics RDP gateway. If you want to use x2go or RDP and have environment modules (see below) that you want to use, then you'll need to run "source startmodules" from your terminal session in the virtual desktop to get them working. ssh is the ideal way to connect if you don't need graphical output on your screen, and that's how most people use the cluster. Please don't run really CPU/memory intensive programs on glamdring itself (unless they're short). Those should be run on the queue ideally. Glamdring is meant for the running of multiple programs in parallel on compute nodes via the queueing system. Running a single program directly on it will not be any faster than running it on your desktop. If you want to run a "normal" (ie. non-MPI, non-OpenMP) program lots of times with different parameters, ( a lot of you do exactly that ) then I have a program which can make that pretty easy - "multirun". More info about it is available here or please email me. It's easier than converting your current "normal" program to MPI. There are various scientific libraries/bits of software installed but you can always ask for whatever you want on there. You can get an account if you're in the Astro department and your supervisor says you can have an account, so get in touch with them or contact Jonathan Patterson ( jop@astro.ox.ac.uk ) direct to setup an account. Below is a guide on how to use the cluster. |
HARDWARE
The machine lives at the Begbroke Science Park and the OS used is Ubuntu 18.04.
Each node is connected by standard gigabit ethernet for normal network tasks like NFS (over which your home directories are connected), and for MPI messages if you're running a parallel MPI program. The berg nodes (more on them later) use a fast, Infiniband network, and everything else is connected over standard gigabit ethernet.
FILE STORAGE
We've got 56TB of disk storage kept in a ZFS array for your home directories. This is backed up twice a month to another storage array kept in a different building. There's also a daily incremental backup, and "snapshots" are taken every hour. So if you need a copy of a file that you've accidentally changed/deleted, I can usually find the version you would like to recover.
Please avoid storing huge numbers of files in a single directory. It might seem easy and convenient for your program to store half a million files in a directory but doing anything in the directory will become very very slow. Avoid anything more than a thousand.
Aside from that, there are various other storage areas which are owned by different groups within Astro, and the 262 TB distributed filesystem /mnt/extraspace which anybody can use. This is the ideal area to use for heavy disk IO since the files are held on multiple compute nodes, which leads to parallel filesystem access. However, it is not backed up, so keep your important files in your home directory.
TRANSFERRING FILES TO THE CLUSTER
I suggest using rsync or scp to transfer files - these are available on linux/OS X and there are scp clients for windows. Typical usage would look like:
rsync -axv localDirectoryName/ myusername@hydra.physics.ox.ac.uk:remoteDirectoryName/
scp -rpv localDirectoryName/ myusername@hydra.physics.ox.ac.uk:remoteDirectoryName/
To transfer the contents of a directory (and any directories in it) over to a subdirectory of your home directory. Feel free to ask me if you have any questions about transferring files.
Transferring back would look like:
rsync -axv myusername@hydra.physics.ox.ac.uk:remoteDirectoryName/ localDirectoryName/
GPUs
Attached to this cluster are various machines for GPU work. The command showgpus will list which nodes have GPUs, what models they are with how much memory, which ones are currently available to be used, and which queue they are in.
Please feel free to ask me if you have problems getting things going, as it can be a bit tricky. To make sure that you get a whole GPU card to yourself on these, please submit the job as:
addqueue -q gpuQueueYouChose -s --gpus numberOfGPUsYouWant --gputype optionallySpecifyTheType -m CPURamYouNeedInGB./yourProgram,
eg. addqueue -q gpulong -s --gpus 1 --gputype rtx2080with12gb ./myProgram
or
addqueue -q gpulong -s -m 4 --gpus 1 ./myProgram
if you don't mind which GPU you get.
COMMONLY USED COMMANDS
There are some commands which are specific to the queueing system. These are detailed below but they are also available in a one-page cheat sheet for your referral later on.
COMPILING
The GNU compilers are installed by default (no environment module needed)
If you're compiling an MPI program, the following commands will call the compiler, linking in the necessary MPI libraries (once you have the openmpi module loaded, as mentioned in SOFTWARE LIBRARIES below):
mpicc, mpicxx, mpif90.
SOFTWARE LIBRARIES
There are lots of libraries/programs available. We use the "Environment Modules" software to manage which ones you want to be available, so:
module avail lists what software modules are available
module list shows which ones you have loaded
module load moduleName loads a module,
eg. "module load openmpi" selects the OpenMPI library
"module load fftw/2.1.5" would load version 2.1.5 of the fftw libraries, and just "module load fftw" would load the default version.
module unload moduleName removes a module
module initadd/initrm/initlist adds/removes/lists which modules are loaded each time you login. So if you use something all the time, add it using that.
Numerous other libraries are available - "module avail" to list them, so have a look in there to see if the library you want is available, but if not I can install it.
Feel free to email me (jop@astro.ox.ac.uk) if you have any problems compiling.
PYTHON LIBRARIES
The OS has python3 installed. You're bound to want to install modules, however. Most python modules are available using "pip", and you would install them with "pip install numpy" for example. This would install them system-wide, however, which you will not have the filesystem permissions to do. You can install any modules you want in your home directory, however, by adding "--ignore-installed --user", so eg. "pip install --ignore-installed --user numpy" would install numpy in your home directory.
Sometimes people need to keep multiple python environments. For example, project1 may need a specific version of numpy but project2 needs another version. You can keep separate python environments with their own modules using a virtualenv. There are numerous tutorials on this out there, and the homepage for virtualenv is https://virtualenv.pypa.io/en/latest/ . This is a good way of managing more complicated python setups.
If you have any problems with any of the above, just let me know and I can help.
USING MPI
MPI is "Message Passing Interface" - library functions for sending & receiving messages between processes. Typically, programs use these to exchange information about which process should be doing what to what - distributing the work.
The default mpi module (which is openmpi), works on both the gigabit ethernet and infiniband (the berg queue) nodes without having to recompile a different version for each network. So compile with that module loaded, and it will work on all the nodes. The berg network is ~ 30 times faster than the normal network, so if you need the extra speed for your MPI communication, please use the "mpi/berg" or "mpi/berg-intel" module instead. Please only use the berg queue if you need that speed, or all the other nodes are busy, because some people can only run on those nodes.
THE JOB QUEUE
We use the slurm queueing system, with some extra queueing/display software I've written which runs on top of it in an effort to make sure everyone gets a fair share of time on the cluster. The queueing on Glamdring is a little complicated since most machines are reserved for the people who bought them. In essence, you can use any "spare time" on them but your compute job will be stopped if an "owner" of the queue needs to use it. You can use the '-i' option to addqueue to get sent an email if this happens, if you like.
If you are part of the cmb or berg groups, then you can use any of the cmb or berg groups - the groups share compute nodes. However, if your job does not use a lot of MPI communication, please use the cmb nodes in preference - the berg ones have a much faster network connection which is needed for lots of the berg simulations.
There are 6 queues you can submit jobs to - "default", "cmb", "blackhole", "cbass", "berg" and "planet". The method below shows how to submit to the default queue (which anybody can use and your jobs will not be stopped) but you can submit to the other queues with the "-q" option to addqueue, eg. addqueue -q cmb
The berg compute nodes have dual infiniband connecting them, so if your job needs fast MPI communication, please use this queue. The berg queue is also limited to 112 cores per person to share it out.
"n -t" will show you what compute nodes there are, how much memory they have, and how busy they are.
MEMORY
Jobs are allocated 0.1 GB of RAM unless you ask for more with the -m flag to addqueue (see below). If you exceed the memory limit, you'll get a message in the job's output file to say so, though sometimes it will just say "Killed.". You can check how much memory it used with the q command.
SUBMITTING YOUR JOB TO THE QUEUE
You can use q -tw 1 to give a list of the compute nodes and how many cores/GB they have free right now. This will tell you what kind of job it's possible to run right now.
addqueue -c "runtimeEstimate or comment" ./myProgramName myParameters
You can just type "addqueue" to get the different options, but here are some common ones:
eg.
addqueue -c "2 days" -n 2x8 ./doSomeAnalysis
will submit your program "doSomeAnalysis" to the default queue, requesting 2 compute nodes with 8 cores on each (so 16 processes are started). You can also use the runtimeEstimate to add your own comments about the job - parameter numbers, maybe, to keep track of which job is which.
addqueue -c "1 week" -m 3 ./doSomeAnalysis
would run a single-process (non-MPI) job with 3GB RAM allocated to it
OR
addqueue -c "1 week" -n 31 ./doSomeAnalysis
would submit a 31-core job to the queue with no mention of how many cores to run on each compute node - so they'll be allocated wherever the queue sees fit. It only really matters how the cores are allocated if your job uses a lot of MPI communication, in which case you'll want as many of them as possible running on each compute node because if the MPI traffic has to go over the ethernet it will be quite slow.
Once you've submitted it you'll see the job number which was allocated to it.
OpenMP
If you are using OpenMP then, to make sure your program runs happily alongside others, you should use the -s option for addqueue, as below. This will set the OMP_NUM_THREADS environment variable so that OpenMP uses the correct number of threads.
addqueue -s -c "test" -n 1x8 myProgram myProgramArguments
which would tell OpenMP to use 8 cores.
If in doubt, feel free to ask me.
Mixing OpenMP and MPI
If you want to use a mix of OpenMP and MPI, here's one way of doing it. You can make a script called, eg. myrun.sh, containing:
#!/bin/bash
export OMP_NUM_THREADS=4
/usr/local/shared/slurm/bin/srun -n 6 -m cyclic --mpi=pmi2 myProgram myParameters
Then make it executable, eg. chmod u+x myrun.sh, and submit it with addqueue -s -n 2x12 -q cmb ./myrun.sh
All this would, for example, allocate 2 nodes with 12 cores each to run the job, then start 6 processes spread over the 2 machines, with each using 4 cores.
Mathematica, Matlab
which matlab or which mathematica will tell you where the actual program is, then you can use that in:
addqueue -n 1x3 -l -s /pathtoprogram/math -script scriptname.m
or
addqueue -n 1x3 -l -s /pathtoprogram/matlab -r functionName
Would reserve 3 cores on the same compute node, and only run 1 occurance (-s) of mathematica at a reduced priority (-l), telling it to run your script scriptname.m
With Matlab specifically, I've seen cases where using parfor or allowing matlab to use all the cores available actually slows the code down, or at least runs at the same speed. So it's worth trying with the -singleCompThread option to matlab. If this runs at the same speed as without it, then please use it, otherwise matlab will use more cores than it needs to. Let me know if you'd like some help with this. If you're using the parpool to run in parallel, and will be running multiple jobs at the same time, please be aware of this likely problem.
CHECKING YOUR JOB
The command q will list your jobs in the queue.
If you have X-windows forwarding enabled, or are using an RDP connection to hydra, then you will get a graphical GUI showing you the jobs. There are various buttons to play with, and the main list of jobs shows all sorts of job info, including current usage stats for the jobs - how many cores, memory GB, diskIO, etc it's using right now. |
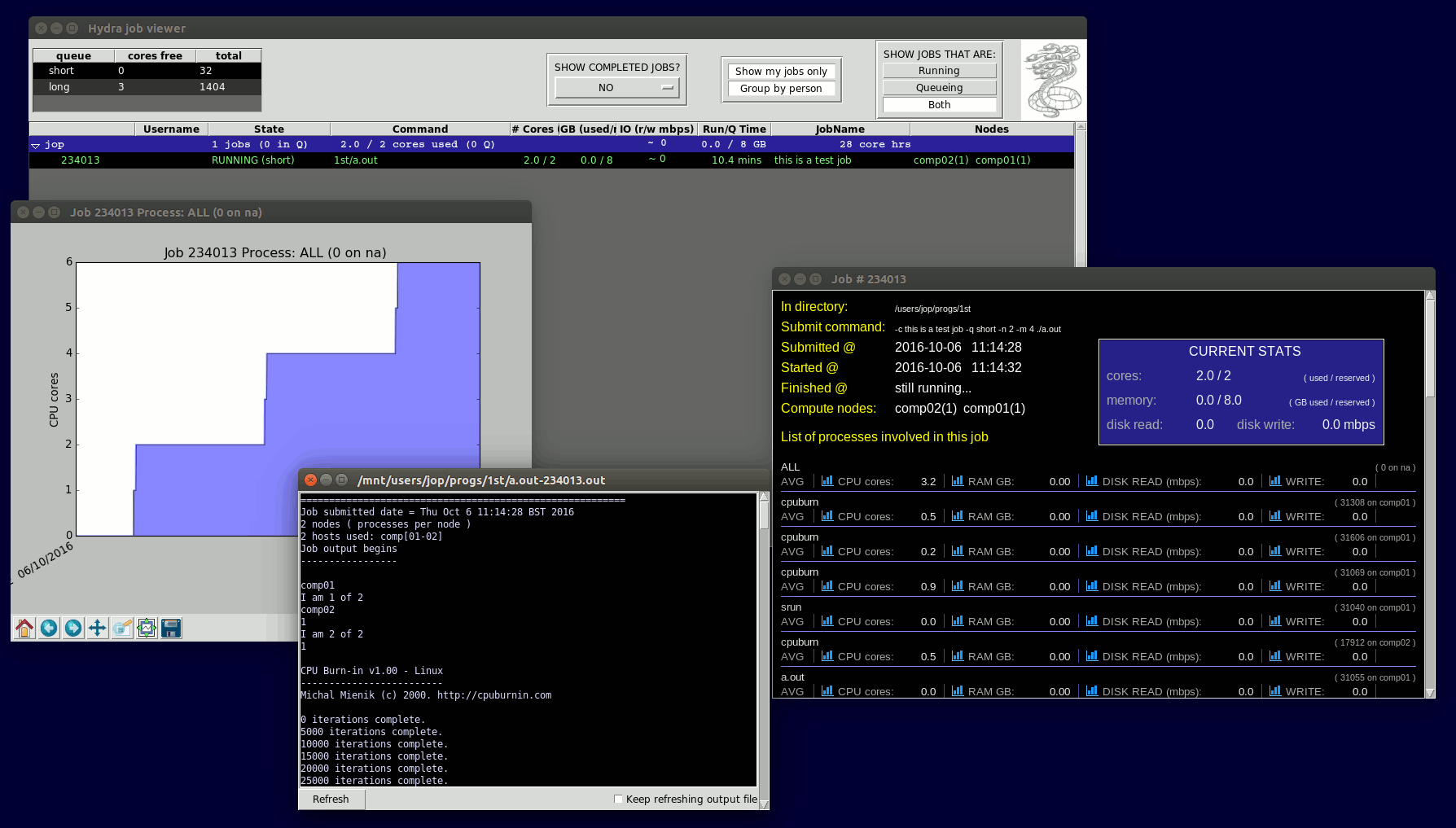 |
You can then double-click on a job to show the job's output file, or right-click to get various options, eg. show job statistics, cancel job, etc
If you select "show job stats", then you will get a window showing CPU, MEMORY, DISKIO usage, and you can click on the icons there to graph these over time. You can use this to check that your job ran efficiently, had enough memory, etc.
If you don't have X-windows on, or you type "q -t", then you'll get a text-only version.
You can check the output (stdout,stderr, or "what would have been printed to the screen") of your job by typing:
q -s jobNunber or (text-only)
showoutput jobNumber
You can stop your job with scancel jobnumber at any time if you want, or via the q GUI interface.
You can also do scancel -u myUserName to cancel all your jobs.
If your job runs longer than you estimated, please update your estimated runtime with the comment command, eg.
comment jobNumber "New comment"
Feel free to login to the compute node running your job and see how it's doing. Have a look at which compute nodes it's running on and then (to see node 12, for example):
ssh comp12
top
exit
If your job sits in the queue waiting for a long time
You can use the command q -tw to show information on what your job is asking for, which compute nodes match that spec., and how many are currently free. This is a good thing to run if your job hasn't started running for many minutes, to see if you're asking for too many resources (and so would have to wait a really long time for the job to start), or to have a guess at when it may start running. You can also run this with a jobnumber of 1 to see what resources are free on the nodes, perhaps to figure out how best to fit a job into the cluster before you submit it.
So that's the basic info for Glamdring. In practice, people have problems compiling, running, and things break sometimes, so please feel free to email me (jonathan.patterson@physics.ox.ac.uk) if you have any questions / think something is wrong.
| File | Size |
|---|---|
| cluster-cheat-sheet-for-users-49164.pdf | 43.87 KB |
Categories: Astrophysics


{kind=link}